

The Council of the European Union has approved the Commission’s proposal to extend the EU certification period for medical devices!
The new legislation provides notified bodies and manufacturers with additional time to certify medical devices, reducing the risk of shortages and ensuring continued access to medical devices for patients in need.
The extension will provide greater flexibility for industry certification of medical devices while ensuring patient safety. The revised timeline will only benefit devices that are safe and have already taken steps to transition to the Medical Devices Regulation.
The proposed amendment to the Medical Devices Regulation has been formally adopted by both the European Parliament and the Council on March 15th 2023. The Commission, Member States, notified bodies, and the medical industry will continue to work on additional measures to address the structural problems and identify medium and long-term solutions.
The transition to the new Regulations must be the collective priority to safeguard patient safety and foster innovation in Europe. To ensure you understand the main areas being addressed by the Commission, we have summarized both Medical Devices Regulation (MDR) and In Vitro Diagnostic Medical Devices Regulation (IVDR) main areas, timelines, and guidance for implementation.
Manufacturers of Medical Devices: MDR Main Areas Summarized
Under the new Medical MDR, manufacturers of medical devices are now responsible for managing the change to get their products CE-marked for the European market. While the MDR has the same basic requirements as the previous Directives, it is generally more stringent, with an emphasis on a life-cycle approach to safety, clinical data, and post-market monitoring.
Manufacturers must comply with the MDR from May 2021, and it is important to start the transition as soon as possible. The Regulation allows for transitional provisions in cases of a high number of devices on the market, anticipated bottlenecks in reviews by notified bodies, and ongoing interpretation of certain provisions.
An early start is especially important due to expected pressure on notified bodies during the transition period. Manufacturers are encouraged to draw up an action plan and prepare for compliance with the MDR, using resources such as the factsheet and step-by-step guide provided by third parties, including the Competent Authorities for Medical Devices (CAMD).
European Commission Timeline for MDR
From May 2024 on, all devices placed on the market must be in conformity with the updated MDR Regulations. While MDD devices already placed on the market may continue to be available through May 2025.
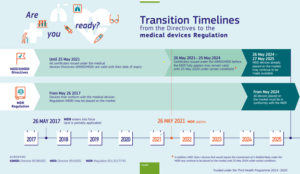
Image source: https://eumdr.com/
Summarized Guidance for Stepping through Implementation for MDR
- Pre-Assessment – ensure clear understanding and the importance of all business implications of the updated MDR. Now is the time to consider any roadblocks or organizational challenges, do you have the staff capabilities or availability?
- Gap Analysis and Actions – First, assess the impact on your product lines and associated internal resources, including existing technical documentation and product labeling, including translation. Determine if changes have impact on budget. Understand the MDR Classes and confirm the conformity routes for all existing and planned future products. Prepare for quality management system (QMS) upgrades including the adequacy of risk management.
- Quality Management System (QMS) – Review that your QMS meets standards and processes for medical devices under the updated regulation and build in the new requirements. Then identify the persons responsible for regulatory compliance within your organization an ensure they are adequately trained and qualified.
- Legal Entities – You should clarify how the company is affected with legal entities organizational structures and resources. Understand product liability and insurance is adequate.
- Portfolio – Complete a cost-benefit analysis for the entirety of your product portfolio, including costs for the potential upgrade of risk class, new procedures for conformity and the costs for post-market surveillance.
- Implementation Plan – Build your roadmap for implementation, including any sub-projects, such as hiring a professional language services provider that meets QMS standards to ensure compliance on documents such as technical documentation and labeling for patient safety. This should also include an appropriate time to train staff on MDR implementation and transition.
- Notified Bodies – Determine the notified bodies capacity and availability to service your implementation plans.
- Execute Implementation Plans – Ensure a cross-functional project management team is in place to cover all aspects of implementation as well as all individual responsibilities have been established.
- Review Efficiency and Effectiveness – Regular meetings are recommended to discuss project status and progress, understand discrepancies, perform gap analysis, risks, next steps and requirements.
- Notified Body Submission – Discuss submission dates to avoid delays in the approval process.
- Ongoing Monitoring – Actively monitor the still-developing European regulatory environment and guidelines expected in the coming months and regularly review the MDR implementation plan, identifying and addressing key areas of risk.
For more, you can visit the European Commission Site.
Manufacturers of In-Vitro Diagnostic Medical Devices: IVDR Main Areas Summarized
Just like the current regulatory regime, manufacturers of in vitro diagnostic medical devices are responsible for obtaining CE-marking for their products for the EU market. However, under the new Regulation, they must manage the transition to obtain CE-marking for their products.
The IVDR has the same basic requirements as the previous Directive. However, it is generally more rigorous, particularly regarding risk classes and oversight provided by notified bodies (NBs). There is also an increased emphasis on a life-cycle approach to safety, supported by clinical data and post-market monitoring such as ‘vigilance’ and ‘post-market surveillance.’
Manufacturers must comply with the IVDR from 2022, and it’s important to start the transition as soon as possible. If necessary, the Regulation allows for transitional provisions due to the high number of devices on the market, anticipated bottleneck in reviews by notified bodies, and ongoing interpretation of certain provisions of the Regulations.
Starting the transition early is especially important due to expected pressure on notified bodies during the transition period. Therefore, manufacturers should approach NBs as soon as possible and check their intentions to apply for designation under the new Regulation, including the scope they intend to cover and when they will be ready.
To comply with the Regulation, manufacturers can utilize the information and links provided below to create an action plan and review the classes their devices fall into under the IVDR.
European Commission Timeline for IVDR
From May 2024 on, all devices placed on the market must be in conformity with the updated IVDR Regulations. While IVDD devices already placed on the market may continue to be available through May 2025.
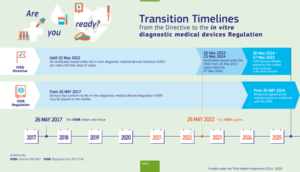
Image source: https://eumdr.com/
Summarized Guidance for Stepping through Implementation for IVDR
- Pre-Assessment – Management should be briefed to understand the importance of business implications of IVDR. Now is the time to consider any roadblocks or organizational challenges, do you have the staff capabilities or availability?
- Gap Analysis and Actions – Asses IVDR implications of product lines, internal resources, and the organization, including budget. Review changes that must be made to existing technical documentation and labeling, including translation. Determine if changes have impact on budget. Understand the IVDR Classes and confirm the conformity routes for all existing and planned future products. Prepare for quality management system (QMS) upgrades including the adequacy of risk management. Prepare for post-market surveillance and post-market performance follow-ups and ensure the respect of traceability of obligations.
- Quality Management System – Review that your QMS meets standards and processes for IVDs under the updated regulation and build in the new requirements. Then identify the persons responsible for regulatory compliance within your organization and ensure they are adequately trained and qualified.
- Legal Entities – You should clarify how the company is affected with legal entities organizational structures and resources. Understand product liability and insurance is adequate
- Portfolio – Do a cost/benefit analysis for your product portfolio; bear in mind costs related to the new risk classification system and the need of involving a Notified Body and costs for post-market surveillance and gaps in the technical documentation, and plan your transition to the IVDR accordingly. This step should include reviewing supply chain provisions, clarify roles and responsibilities of business partners.
- Implementation Plan – Build a roadmap for implementation, including definition of sub-projects, resource requirements and a steering group, and ensure overall responsibility for IVDR implementation has been established. Give special consideration to certificate expiration dates bearing in mind the translational period, provisions, and availability of your notified bodies. Plan for training and workshops for IVDR implementation.
- Notified Bodies – Determine your notified bodies capacity and availability to service the implementation plan.
- Execute Implementation Plan – Implement all sub-projects to ensure compliance, including partnering with a professional language services provider. Ensure all cross-functional project management team is in place to cover all aspects of implementation and all individual responsibilities for IVDR implementation have been established.
- Review Efficiency and Effectiveness – Hold regular meetings and progress reviews on project status and progress, discrepancies and gap analysis, assess risks, next steps and requirements.
- Notified Body Submission – Discuss submission dates to avoid delays in the approval process.
- Ongoing Monitoring – Actively monitor the still-developing EU regulatory environment and guidelines expected in the coming months and establish a procedure for dealing with unannounced inspections from notified bodies. In addition to regularly reviewing IVDR implementation plans and addressing key areas of risk.
For more, you can visit the European Commission site.
Considerations for Translations in the EU MDR & IVDR
EU MDR and IVDR involve the translation of products into the 24 official languages, depending on the product’s presence. To ensure compliance, it is recommended to work with an LSP that specializes in regulatory submissions, packaging, labels, IFUs, manuals, and software. Comprehensive preparation for the translation process can lead to cost and time efficiency, including considering multi-language translation, shared content, standardized formats, and leveraging translation memories.
Morningside, a Questel Company, is hosting a webinar on March 23 from 1 PM-2 PM with MDx CRO to discuss the challenges medical device organizations face in compliance with MDR and IVDR regulations, including translations. The webinar will feature two expert speakers from the regulatory and translation industries, Carlos Galamba and Anna Eisenberg, who will provide insights into the latest amendments and cost-effective language translation approaches.
Carlos Galamba is a seasoned professional in the diagnostics industry and the co-founder of MDx CRO, and Vice President for Diagnostics at RQM+. Anna Eisenberg is an Associate Director of Life Science Business Development at Morningside with 20+ years of experience in the localization industry and a focus on life sciences.
About Morningside
Morningside, a Questel Company, provides life sciences organizations with end-to-end language solutions for their regulatory, clinical, commercial, and patent needs. With over 4,000 clients in 55 countries, they are recognized for their life science expertise and technology innovation across pharma, biotech, medical devices, and healthcare. Their life sciences translation services ensure products and ideas reach new markets and comply with all regulatory and cultural requirements throughout the product lifecycle.
Those interested can register for the free webinar “Meeting MDR & IVDR Regulations: Translation & Cost-Savings” on March 23rd from 1 PM-2 PM EST. If unable to attend at that time, the recorded webinar will be shared with registrants post-event. The webinar can save time and ensure compliance in the organization while building a reputation in the medical device industry.
References:
Getting ready for the new regulations. European Commission. (n.d.). Retrieved March 10, 2023, from https://health.ec.europa.eu/medical-devices-new-regulations/getting-ready-new-regulations_en
Press corner. European Commission. (2023, March 7). Retrieved March 16, 2023, from https://ec.europa.eu/commission/presscorner/detail/en/STATEMENT_23_1504
Manufacturers MD. European Commission. (n.d.). Retrieved March 10, 2023, from https://health.ec.europa.eu/medical-devices-topics-interest/reprocessing-medical-devices/manufacturers-md_en
Manufacturer IVD. European Commission . (n.d.). Retrieved March 10, 2023, from https://health.ec.europa.eu/medical-devices-new-regulations/getting-ready-new-regulations/manufacturer-ivd_en

Madeline Sturdivant, Brand Marketing Manager at Morningside, a Questel Company, is an experienced marketing professional with five years in multiple disciplines, including digital, print, event, and strategy. She is responsible for the company's global marketing activities and managing relationships with the world's largest and most demanding healthcare, legal, and corporate organizations. Madeline's varied personal and professional interests drive her to continually expand her knowledge to benefit those around her.